Article Text

Statistics from Altmetric.com
Introduction
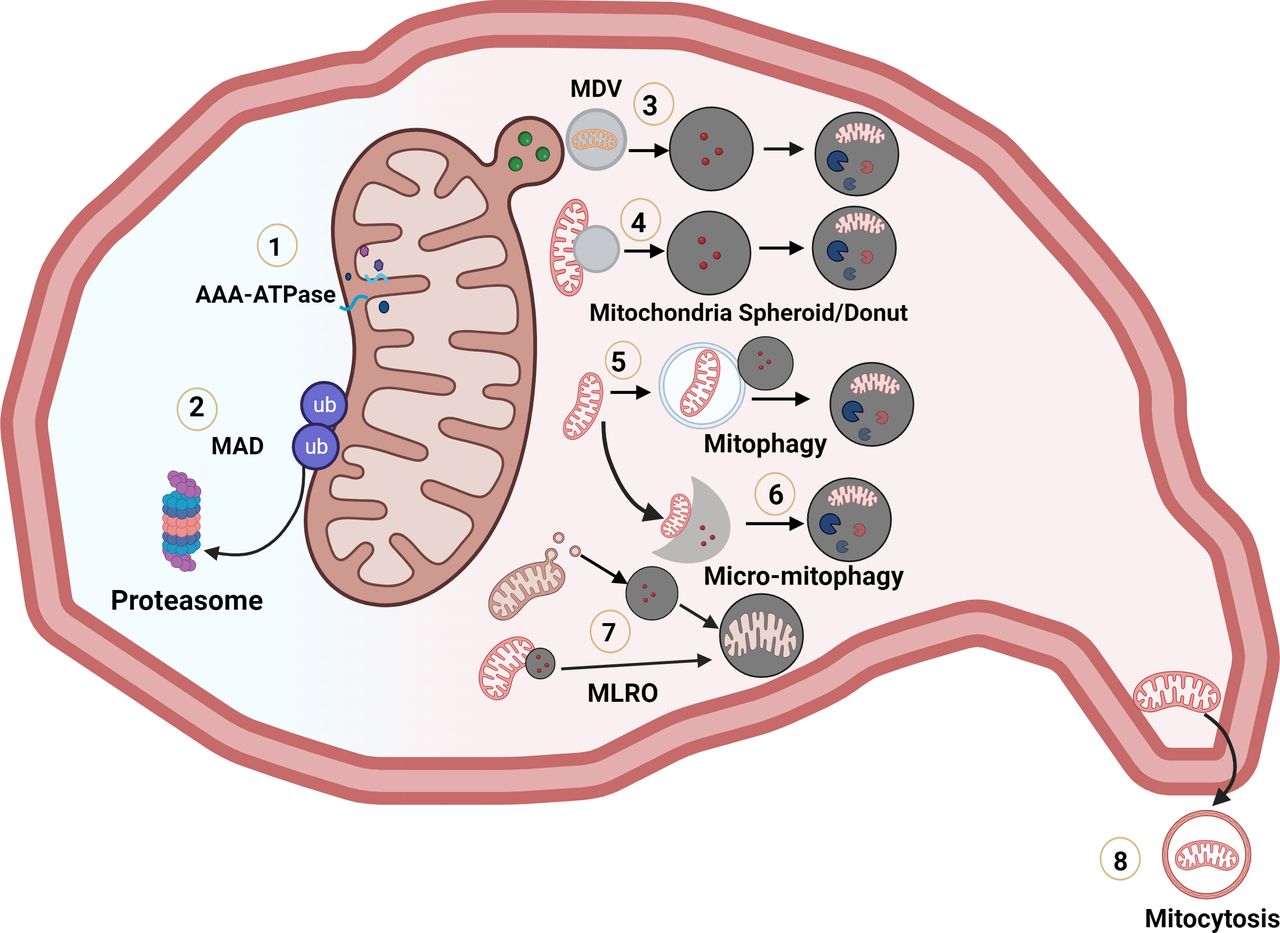
According to the widely accepted ‘endosymbiotic theory’, mitochondria are originated from bacteria when the eukaryotic cell evolved from the capture of alphaproteobacteria, within primitive archaea around 2 billion years ago, which set a symbiotic relationship between the captured bacterium and its archaeal host. Mitochondria are double membrane-bound organelles that have unique sub-compartments: outer mitochondrial membrane, inner mitochondrial membrane, inter-mitochondrial membrane space and mitochondrial matrix. Mitochondria are not only the ‘powerhouses’ for generation of ATP for cellular functions and survival but also are critical for lipid and glucose metabolism, calcium buffering, phospholipid synthesis, programmed cell death and innate immune response. Given their crucial roles in maintaining cellular functions, cells have developed intricate mechanisms to regulate mitochondrial morphology, quality and numbers.1–5 Disruption of mitochondria functions has been implicated in various liver diseases including alcohol-associated liver disease (ALD), metabolic dysfunction-associated fatty liver disease (MAFLD), drug-induced liver injury, chronic viral hepatitis, liver cirrhosis and hepatocellular carcinoma (HCC).6 Below we summarise eight distinctive mechanisms that regulate mitochondria quality and turn over (figure 1), in particular, the newly discovered mitochondria-lysosome-related organelle (MLRO) in liver biology and liver diseases.

Multiple pathways regulating mitochondrial quality control and turnover. (1) Degradation of mitochondria matrix, inner membrane and intermembrane oxidised or misfolded protein by mitochondrial proteases. (2) Ubiquitinated outer mitochondrial membrane proteins are extracted and degraded by the ubiquitin-proteasome system. (3) A portion of mitochondria budding off as mitochondria-derived vesicles and fuse with lysosomes for degradation. (4) Depolarised mitochondria undergo morphological remodelling to squeeze their matrix to form donut-shaped mitochondrial spheroids and acquire lysosomal contents for possible degradation. (5) Canonical autophagosomes envelop mitochondria for lysosomal degradation via PINK1-PARKIN dependent or independent manner. (6) Lysosomes directly take up mitochondria by the lysosomal membrane invagination. (7) Endosome/pre-lysosome directly ‘hit and run’ with mitochondria or MDV to form mitochondria-lysosome-related organelle (MLRO). (8) Mitochondria are secreted from cells by migrasomes or autophagic secretion of mitochondria. Figure 1 was Generated using BioRender. MAD, mitochondrial-associated degradation; MDV, mitochondria-derived vesicle.
Mitochondrial ATPases associated with diverse cellular activities (AAA ATPase)-mediated degradation
A mitochondrion contains around 1500–2000 proteins, with over 99% being coded in the nucleus and imported from the cytosol. Only 13 proteins are coded by the mitochondrial genome.7 Both imported proteins and mitochondrial encoded proteins have to be properly assembled to form complexes for proper mitochondria functions. Mitochondria are also the major site to produce reactive oxygen species that can oxidise and damage mitochondrial proteins, and levels of oxidised mitochondrial proteins often increase with ageing. Thus, mitochondria have their own AAA ATPase family proteases located on both sides of the inner membrane, which are responsible for the degradation of the unfolded or oxidised proteins in matrix and intermembrane space.8 Failure to degrade these oxidised and damaged proteins timely may lead to mitochondrial dysfunction. As a result, a number of human diseases are associated with impaired or improper mitochondrial protein degradation, including cancer and numerous neurodegenerative diseases. However, studies on the role of mitochondrial AAA ATPase in liver pathobiology have been scarce, which will be an exciting future field to explore.
Mitochondrial-associated degradation (MAD)
The endoplasmic reticulum (ER) is a major site for protein folding to ensure proper protein assembly and trafficking of most secretory and membrane proteins. To ensure the fidelity of protein folding and maturation, cells have developed delicate quality control mechanisms to timely remove misfolded proteins, and these mechanisms include unfolded protein response, ER-associated degradation (ERAD) and autophagy.9 10 Misfolded ER membrane proteins are ubiquitinated by the E3 ligase hydroxymethyl glutaryl-coenzyme A reductase degradation protein 1 with its co-factor protein sel-1 homolog 1, and then are proteolytically degraded by 26S proteasome.9 This degradation is coupled with a membrane extraction process governed by AAA ATPase Valosin-containing protein (VCP/p97, Cdc48 in yeast) and its cofactors.11
Similar to the ERAD process, some outer mitochondrial membrane (OMM) proteins can be ubiquitinated and degraded by the ubiquitin-proteasome system, which is termed as ‘MAD’.4 During MAD, aberrant ubiquitinated OMM proteins are extracted by type II AAA ATPase VCP/p97 to facilitate the degradation by the proteasome. MAD has several substrates, including mitochondrial fusion proteins (mitofusin 1 and 2), anti-apoptotic Bcl-2 family protein (MCL-1), ER-mitochondria contact site protein (Mdm34p) and mitochondrial protein import (MSP1P, TOMM70).4 It is worth noting that the MAD pathway has been extensively studied in yeast and cultured mammalian cells. However, its relevance to liver pathophysiology is still not well understood, despite a study showing that increased VCP/p97 expression is connected to increased HCC recurrence.12 To further understand the role of MAD in liver diseases, it is necessary to develop relevant mouse models and conduct more investigations in the future.
Mitochondria-derived vesicles (MDVs)
MDV was originally discovered by McBride’s group. When mitochondria-anchored protein ligase was overexpressed in cultured cells, they observed vesicular structures with diameters of 70–100 nm derived from mitochondria and they coined the name MDVs for these structures.13 Stress-induced MDVs are selectively enriched with oxidised mitochondrial proteins and are degraded within lysosomes following MDV-lysosome fusion, suggesting that MDVs may constitute an alternative mitochondrial quality control mechanism.14 15 Indeed, MDVs have been shown to protect against hypoxia-induced cardiomyocyte damage and to be involved in mitochondrial antigen presentation as an immune response pathway provoked by inflammation during Parkinson’s disease pathogenesis.16 However, the role of MDVs in liver pathophysiology remained to be determined.
Mitochondrial spheroid
Damaged mitochondria undergo dramatic structural remodelling to form mitochondrial spheroids serving as another quality control mechanism.17 18 Induction of mitochondrial spheroids are independent of autophagy-related 5 (ATG5) and ATG7 and is negatively regulated by PARKIN.17 18 Mitochondrial spheroids have a ring or cup-shaped morphology with a squeezed mitochondrial matrix. Interestingly, they also enclose intracellular organelles such as ER, lipid droplets, other mitochondria and cytosolic proteins. Mitochondrial spheroids have been observed in acetaminophen-induced liver injury and in alcohol-fed mouse livers.18–20 It is still unclear whether the contents acquired by mitochondrial spheroids, which include some lysosome proteins, are degraded within the mitochondrial spheroid lumen. Moreover, whether and how mitochondria spheroid would contribute to drug-induced liver injury and ALD requires further investigation in the future.
Macroautophagy-mediated removal of mitochondria (mitophagy)
Mitophagy is generally defined by the selective enveloping entire dysfunctional/excess mitochondria by autophagosomes and trafficking to lysosomes for degradation in either PINK1-PARKIN dependent or independent manner.3–5 The mechanisms and pathophysiological role of mitophagy in human diseases, including liver diseases, have been extensively reviewed,3 5 6 21 which will not be discussed here.
Micromitophagy
While macromitophagy involves the autophagosome-mediated wrapping of mitochondria, lysosomes can directly take up various types of cargoes, including mitochondria, by enfolding the lysosomal membrane. This leads to the degradation of mitochondria within the lysosome, a process termed as micromitophagy.22 Unfortunately, there is very little research on micromitophagy compared with macromitophagy. At present, the mechanisms that regulate micromitophagy and how it contributes to liver diseases are mostly unknown.
Mitocytosis and autophagic secretion of mitochondria (ASM)
Migrasomes are large vesicular structures grown on the retraction fibres during cell migration and function as organelles for non-autonomous cell–cell communication.23 Migrating cells transport damaged mitochondria into migrasomes for disposal, a process called mitocytosis, an alternative mitochondrial quality control mechanism independent of autophagy.24 In addition, mitochondria can be secreted through a process called ASM, which is distinct from mitocytosis. ASM operates independently of ATG7-mediated autophagy but requires ATG14-FIP200 and SNAP23-mediated secretory autophagy. In addition to clearance of damaged mitochondria, ASM may activate the innate immune cGAS-STING pathway in the recipient cells.25 No studies have been conducted to characterise the role of mitocytosis and ASM in liver disease. However, it is highly likely that they are involved in many chronic inflammatory liver diseases.
Mitochondria-lysosome-related organelle
The liver is a central organ that regulates the metabolism of carbohydrates, lipids, proteins and amino acids and also plays a major role in the processing and elimination of drugs and toxins from the body. Hepatocytes are the main parenchymal cells of the liver, accounting for approximately 80% of its volume. However, when primary hepatocytes are cultured in vitro, they tend to rapidly lose their specific functions, resulting in dedifferentiation, morphological changes and loss of metabolic enzymes. This dedifferentiation process also occurs in vivo, leading to reduced synthetic liver capacity and promotes chronic liver diseases such as ALD and metabolic dysfunction-associated steatohepatitis (MASH).26 27
In a recent study published in Cell Reports, we discovered MLRO as a hybrid cellular organelle that contains both mitochondrial and lysosomal markers in primary cultured hepatocytes undergoing dedifferentiation.28 MLRO is an electron-dense organelle that has either single or double membrane containing undegraded electron-dense onion-like membranes with other heterogeneous contents (figure 2A). MLRO has an acidic luminal pH and is either derived from MDVs that fuse with lysosomes or from direct ‘hit and run’ of endosomes/pre-lysosomes with mitochondria. Induction of MLRO is independent of canonical autophagy (ATG5) and mitophagy machinery (PARKIN). Moreover, the number of MLROs is correlated with mitochondria degradation, suggesting that MLRO is an alternative mitochondrial quality control mechanism independent of canonical mitophagy. From an economic perspective, inducing MLRO is more efficient and faster than canonical mitophagy that requires the formation of double-membrane autophagosomes to surround mitochondria. Our findings also show that inducing MLRO rather than traditional mitophagy is the primary pathway for degrading mitochondria in dedifferentiated hepatocytes. Hepatocyte dedifferentiation is very common in the late stage of chronic liver diseases such as ALD and MASH, which leads to liver failure. Indeed, we found that increased MLRO is also readily observed in diet-induced MASH in mice, implicating the relevance of MLRO in liver diseases.

{kind=link}
{kind=link}
TFEB and MLRO in hepatocyte dedifferentiation and alcohol-associated hepatitis (AH) and metabolic dysfunction-associated steatohepatitis (MASH). (A) The images show MLRO in primary cultured mouse hepatocytes. The bar scale is 500 nm. (B) The proposed model illustrates how lysosomes and TFEB regulate MLRO in hepatocyte dedifferentiation. Increased amino acid levels from lysosomal degradation promote mTOR translocation to the lysosomal membrane through the ‘Ragulator’ complex, leading to mTOR activation. This, in turn, phosphorylates TFEB, causing TFEB cytosolic retention and inactivation. When amino acid levels from lysosomal degradation are low, mTOR is inactivated, leading to decreased TFEB phosphorylation and increased nuclear TFEB translocation. TFEB then binds to Coordinated Lysosomal Expression and Regulation (CLEAR) motif-containing target genes involved in autophagy and lysosomal biogenesis. TFEB-mediated lysosomal biogenesis promotes the clearance of MLRO, inhibits hepatocyte dedifferentiation and attenuates the pathogenesis of AH and MASH. Figure 2B was generated using BioRender. MLRO, mitochondria-lysosome-related organelle; TFEB, transcription factor EB.
Since the rate of mitophagy, mitochondrial spheroid, mitocytosis and ASM is low in primary cultured degenerated hepatocytes, it is tempting to speculate that MLRO is specifically associated with hepatocyte dedifferentiation and liver degeneration. Mitochondria and lysosomes are two critical organelles that not only regulate nutrient and energy homeostasis by effectively recycling or turning over cellular components but also serve as key hubs of signalling cascades for cell survival and differentiation or dedifferentiation. Notably, MLRO is somewhat similar to the damaged lysosomes as MLRO are galectin 3 positive, a well-known marker for damaged lysosomes. In addition to degradation, lysosomes are also intracellular signalling hubs for sensing nutrients and energy for mTORC1 and AMPK activation. Mouse embryonic stem cells (mESCs) lacking AMPK exhibited severe defects during differentiation, which is associated with decreased transcription factor EB (TFEB),29 a master transcription factor for regulating lysosomal biogenesis. TFEB mutant mESCs and bafilomycin A1-treated mESCs lacking functional lysosomes showed a decrease in the expression of hepatocyte nuclear factor 4α (HNF4α),29 a crucial transcription factor that regulates hepatocyte identity. In another study using a genome-wide screen that sought to identify genes whose deletion suppresses mESCs differentiation, Villegas et al30 identified multiple genes involved in lysosomal responses to amino acid availability that promotes the cytoplasmic retention of transcription factor E3 (TFE3), a homologue of TFEB, as a critical regulator of ESC differentiation. These emerging findings suggest that lysosomal-mediated nutrient metabolism and signalling is critical for TFEB/TFE3 regulation in cell differentiation and dedifferentiation. We also demonstrated that the overexpression of TFEB not only increased the clearance of MLRO but also augmented the expression of HNF4α, which inhibited hepatocyte dedifferentiation. Although it is still unclear how TFEB affects HNF4α expression, these findings suggest a novel concept where alterations of mitochondria and lysosomes could regulate hepatocyte dedifferentiation and thereby contribute to chronic liver diseases such as ALD and MASH.
While induction of MLRO is clearly associated with hepatocyte dedifferentiation, there are several important questions that remain unanswered. The mechanisms that regulate the formation of MLRO are still unclear. The proteins responsible for the fusion of MDVs with lysosomes to form MLRO are yet to be identified. Our observation of ‘hit and run’ events between endosomes/lysosomes and mitochondria suggests that some lysosomal proteins or enzymes may transfer into the mitochondria, which could lead to either the acidification or the degradation of mitochondrial proteins. Alternatively, mitochondria could acquire V-ATPase, resulting in the decrease of mitochondrial pH and acidification, ultimately transforming the mitochondria into MLRO. Further studies are necessary to explore these possibilities.
In summary, MLRO is an alternative mechanism for mitochondrial quality control independent of canonical autophagy/mitophagy. Modulating the formation of MLRO may be important in regulating hepatocyte dedifferentiation, which may be beneficial for chronic liver diseases such as ALD and MASH.
Data availability statement
All data relevant to the study are included in the article.
Ethics statements
Patient consent for publication
Ethics approval
Not applicable.
References
Footnotes
Twitter @Hongmin Ni
Contributors XM, H-MN and W-XD conceived and wrote the manuscript. W-XD made the graphs and figures.
Funding This work was partially supported by NIH grants: R37 AA020518, R01 DK102142 and R01 AG072895 (W-XD).
Competing interests None declared.
Patient and public involvement Patients and/or the public were not involved in the design, or conduct, or reporting, or dissemination plans of this research.
Provenance and peer review Not commissioned; externally peer reviewed.